
Fanconi anemia
Last Updated December 20th, 2021
What is Fanconi anemia ?
Fanconi anemia is a serious blood disorder that is characterized by the inability of the bone marrow to make enough new blood cells. More than 50% of the people diagnosed with this disease get it before the age of 10. This disease is more common in people who have birth defects such as brachydactyly, skin pigmentation defects, microcephaly, microphthalmia and abnormal kidney structures.
This condition often results in the progressive deficiency of red blood cells, white blood cells, and platelets. People with this condition are also at an increased risk of developing bone marrow cancer. Fanconi anemia is equally prevalent in both men and women and in all ethnic groups.
Causes
 Fanconi anemia is a genetic disorder that is inherited. If there are mutations in at least 18 genes, the occurrence of this condition is high. The chromosomes of people affected by this condition are not able to repair the damaged DNA. Hence they break and rearrange easily leading to chromosome instability.
Fanconi anemia is a genetic disorder that is inherited. If there are mutations in at least 18 genes, the occurrence of this condition is high. The chromosomes of people affected by this condition are not able to repair the damaged DNA. Hence they break and rearrange easily leading to chromosome instability.
The majority of the Fanconi anemia cases happen due to mutations in one of these genes, FANCA, FANCC, and FANCG. These genes are responsible for providing instructions for the production of blood cells. Sometimes, the mutation of these genes may also lead to uncontrolled cell growth, which results in cancer.
Autosomal Recessive
This condition is mostly inherited in an autosomal recessive manner. If a person only has one abnormal gene of Fanconi anemia, he/she becomes a carrier, with no apparent symptoms. However, if a person gets 2 abnormal genes, one from each parent, then he/she gets affected by this condition.
If both the parents have the mutated gene related to this condition, the risk of it being passed on to their child (both male and female) is 25%. The risk of the child becoming a carrier is 50%. Lastly, the chance of the child not getting the disorder at all is 25%. If the parents are consanguineous (close relatives), there is a higher chance of their child having and a recessive genetic disorder like Fanconi anemia.
This disorder can also be inherited if there are mutations in the following genes, BRCA2, BRIP1, FANCB, FANCD2, FANCE, FANCF, FANCI, ERCC4, FANCL, FANCM, PALB2, RAD51C, SLX4, and UBE2T.
The mutation of the FANCB gene mostly affects males as it is located in the X chromosome. Females with X-linked recessive trait are usually carriers as they have two X chromosomes. Males have only 1 one X chromosome that is inherited from the mother (fathers pass on the Y genes to males). If the male receives the X chromosome with the altered gene, he will develop this disorder.
Female and male carriers of the disease
 Female carriers of an X-linked disorder have a 25% chance (with each pregnancy) of producing a carrier daughter, 25% chance of producing a non-carrier daughter, 25% chance of producing a son who will be affected by the disease and 25% chance of producing an unaffected son.
Female carriers of an X-linked disorder have a 25% chance (with each pregnancy) of producing a carrier daughter, 25% chance of producing a non-carrier daughter, 25% chance of producing a son who will be affected by the disease and 25% chance of producing an unaffected son.
Meanwhile, if a male with an X-linked disorder reproduces, he will pass on the abnormal gene to all his daughters. He will not be able to pass on the disorder to his son as males only pass on the X chromosome to their daughters.
Lastly, this condition can also be inherited in an autosomal dominant manner. This happens when there are mutations in the RAD51 gene. In dominant genetic disorders, only a single abnormal gene is necessary to cause a particular disease. The abnormal gene can be inherited from either parent or result from a new mutation in the patient. The risk of passing the mutated gene from parent to offspring is 50% in both males and females. People who develop FA due to a mutated RAD51 gene usually have a mutation that occurs in the egg or the sperm. In this instance, FA is not inherited from the parents.
Symptoms
The symptoms of Fanconi anemia are different from person to person. However, it is often characterized by a variety of physical abnormalities, which reveal themselves in early childhood. Some of the symptoms of Fanconi anemia are
- Low birth weight
- Gastrointestinal difficulties
- Skin pigmentation
- Small reproductive organs in males
- Short stature
- Kidney structural problems
- Thumb and arm anomalies
- Defects in tissues separating chambers of the heart
- Small head
- Small, crossed, or widely spaced eyes
 Blood problems start once the individual reaches 6-8 years and they may start experiencing
Blood problems start once the individual reaches 6-8 years and they may start experiencing
- Anemia
- Tiredness
- Sleepiness
- Weakness
- Lightheadedness
- Dizziness
- Irritability
- Headaches
- Pale skin color
- Difficulty breathing
- Cardiac symptoms
The more serious symptoms of this disorder are bone marrow failure and an increased risk of developing cancer.
Bone Marrow Failure
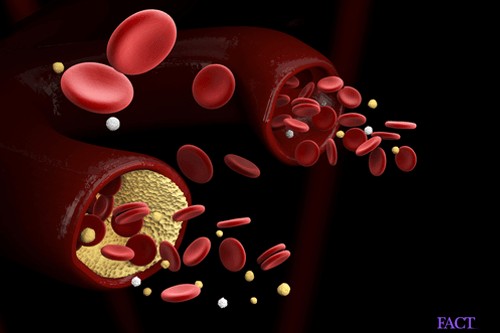
Bone marrow is a semi-solid tissue that is found inside the spongy portions of long bones. The main function of the bone marrow is to produce hematopoietic stem cells that eventually become platelets, erythrocytes (red blood cells) and leukocytes (white blood cells).
Progressive bone marrow failure usually happens before the age of 10. It is characterized by low platelet levels or low white blood cells.
Cancer
People who have Fanconi anemia are at an increased risk of developing certain cancers (acute myeloid leukemia and specific solid tumors)as compared to the general population. Fanconi anemia mainly causes cancer that affects the head and neck region, gastrointestinal tract, esophagus or gynecologic regions.
Who should be tested?
Any infant born with the thumb and arm abnormalities should be tested for FA. Any person who develops aplastic anemia (a conditionwhich results in your body not producing enough new blood cells) at any age should also be tested for FA. People (especially those without a history of tobacco or alcohol use), who develop squamous cell carcinoma of the head neck, gastrointestinal or gynecologic system should also be tested. This is because chemotherapy and radiation may have an adverse effect on FA patients.
Diagnosis
This condition is usually diagnosed by a combination of methods that may include
Clinical evaluation
Although Fanconi anemia patients can be identified with certain physical abnormalities such as small head, deformed thumb, skin pigmentation, the doctors may order the following test to be certain
- Ultrasonography of the kidneys and urinary tract
- Hearing test
- Developmental assessment (especially for toddlers and school-age children)
- Evaluation by a hematologist
- Full blood typing
- HLA typing of the affected individual, siblings, and parents for consideration of hematopoietic stem cell transplantation
- Blood chemistries
- Bone marrow aspiration
Specialized tests
Chromosome breakage test
The widely used test for diagnosing FA is called a chromosome breakage test, which involves treating some of the patient’s blood cells in a test tube with a chemical that crosslinks DNA. DEB (diepoxybutane) and MMC (mitomycin C) are the two chemicals that are commonly used for this test. When the cells come in contact with these chemicals, the DNA breaks up. Normal cells are able to correct most of the damage but cells affected with FA display chromosome breakage. These tests can also be performed prenatally by obtaining cells from the amniotic fluid or the chorionic villi.
Molecular genetic testing
Molecular genetic testing can be done for all 18 genes that are related to FA. Firstly, a complementation test is done to identify why the gene is mutated. Once identified, sequence analysis of the gene is done to determine the specific mutation in that particular gene. If the mutation is not identified, deletion/duplication analysis has to be done for the genes associated with FA.
Treatment
The treatment of FA is done depending on the symptoms of the condition in each patient. Treatment plans usually require coordinated efforts by pediatricians, surgeons, cardiologists, kidney specialists (nephrologists), urologists, gastroenterologists, etc. These are some of the most common treatment methods for FA patients
Androgen administration
The administration of androgens (male hormones) is known to improve the blood count in approximately half of the patients diagnosed with FA. The first signs of this treatment plan will see the increase in hemoglobin, generally after a couple of months following treatment. The red blood cells count also improves. However, it takes time for the white blood cells and platelet count to improve. Also, the body builds resistance to this treatment after a while.
Hematopoietic growth factors
This therapy involves the administration of glycoprotein, a compound that stimulates the bone marrow to produce more granulocytes and stem cells. This improves the white blood cells (mainly neutrophil) count in some individuals. This therapy’s main side effect is that the patient may develop a skin condition called Sweet’s syndrome.
Hematopoietic stem cell transplantation
This therapy involves the transplantation of donor stem cells (preferably from a close relative) obtained from bone marrow, peripheral blood, or cord blood. The stem cells automatically increase the blood count in patients. HSCT remains the only curative treatment for patients with Fanconi anemia.
Surgery
Surgery is sometimes needed to correct the skeletal malformations associated with this disease.
Prevention
This condition cannot be prevented but people with a family history of this disease can undergo genetic counseling before planning to have a baby to understand the risks better. Pregnant women, who have a family history of this disease, should also do a genetic test to determine whether the baby has FA. This can be done via two procedures
Amniocentesis: In this procedure, the doctor uses a needle to remove some amniotic fluid, which is then tested for FA genes.
Chorionic villus sampling: This is done by inserting a tube through the vagina to take tissue samples of the placenta. The samples are then tested for FA genes.
If the baby is tested positive, the doctor will run more tests to determine the severity of the disease. If the disease is too severe, the couple may opt to undergo an abortion.
https://www.nhlbi.nih.gov/health-topics/fanconi-anemia
https://medlineplus.gov/ency/article/000334.htm
https://ghr.nlm.nih.gov/condition/fanconi-anemia
https://medlineplus.gov/ency/article/000334.htm
https://jmg.bmj.com/content/40/1/1
https://www.cincinnatichildrens.org/health/f/fanconi-anemia
https://www.stanfordchildrens.org/en/topic/default?id=fanconi-anemia-160-10
Dos and Don'ts
Dos
- You should be careful not to injure yourself
- You should get your blood count tests done regularly
- You should undergo a genetic counseling session if you are planning to have a baby
Don'ts
- Drink or smoke cigarettes. Nicotine and alcohol may increase the risk of chromosomal breakage in individuals with FA.
- Feel low if you have FA. Improvements in blood and bone marrow stem cell transplants have greatly involved the survival rates of people living with this condition.
- Play contact sports as you may get injured. People with FA should not risk injury as their blood might not clot due to low platelet count.
Help Others Be Fit
Related Conditions





Trending Topics








































































































































About
Related Conditions


Subscribe to free FactDr newsletters.
REVAMP YOUR
LIFE
HEALTH
WELLNESS

If you're enjoying our website, we promise you'll absolutely love our new posts. Be the first one to get a copy!
Get factually correct, actionable tips delivered straight to your inbox once a week.
We hate spam too. We will never share your email address with anyone. If you change your mind later, you can unsubscribe with just one click
By clicking Subscribe, I agree to the FactDr Terms & Conditions & Privacy Policy and understand that I may opt out of FactDr subscriptions at any time.
×
How can we improve it?
×
Happy to know you loved our article!
Did it give you information that you used / can use in your life?

