
Apert Syndrome
Last Updated December 20th, 2021
Overview
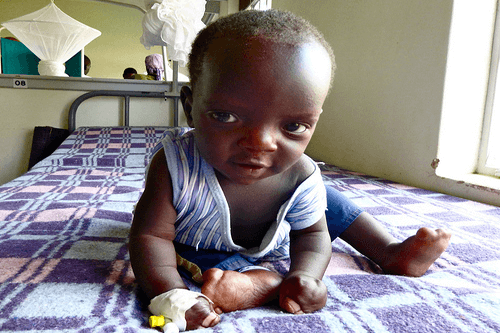
Apert syndrome is a congenital disorder identifiable by birth defects such as an abnormal form of the head and webbed condition of the hands and feet. It is quite rare and estimates of its incidence vary somewhat, with recent studies placing it between 1 in 65,000 and 1 in 88,000 live births.
Asian populations display the highest incidence while Hispanics are found to have the lowest. This disorder can affect both males and females in equal numbers and has no known cure at present. Scientists believe that it arises due to the occurrence of a rare genetic mutation. This means that Apert syndrome is a potentially heritable condition. Yet, in most cases, the mutation arises randomly or ‘sporadically’. Apert syndrome derives its name from that of the French physician, Eugene Apert, who produced detailed case studies of this condition.
What Is Apert Syndrome?
 Apert syndrome is a rare genetic disorder that involves premature fusing of the skull bones during the development of the fetus. This results in a distorted skull shape impairing the normal growth of the brain. As the brain continues to grow within this restricted space, unable to expand normally towards the front and back, it begins to push outwards sideways. This puts pressure on the bones of the skull and face, resulting in further malformations.
Apert syndrome is a rare genetic disorder that involves premature fusing of the skull bones during the development of the fetus. This results in a distorted skull shape impairing the normal growth of the brain. As the brain continues to grow within this restricted space, unable to expand normally towards the front and back, it begins to push outwards sideways. This puts pressure on the bones of the skull and face, resulting in further malformations.
Babies with Apert syndrome also usually display an abnormal fusion of the bones in the hands and feet with noticeably webbed fingers and toes. They may also have extra fingers or toes, although this is rare. Congenital organ defects such as those in the heart can also be present. Since the initial growth of the brain is affected due to abnormal development of the skull, intellectual disability is common. However, cognitive function can also be normal in some cases.
What Is Responsible For It?
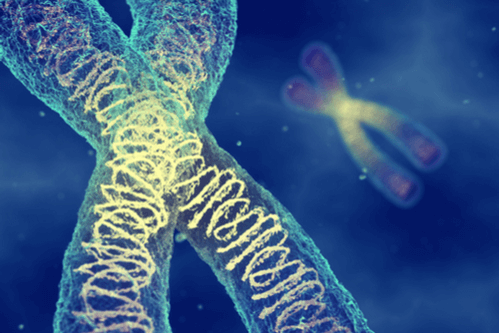
A gene known as FGFR2 is responsible for the formation of bone cells and the subsequent development of bone structure. A certain mutation arising in this gene is found to be the primary cause of Apert syndrome. This mutation could potentially be triggered by environmental factors. It arises spontaneously and randomly in most instances but can also be inherited. There is a 50% probability of an affected individual passing on the defective mutation to their offspring and in such cases, the risk is seen to increase with parental age.
What Are The Indications of Apert Syndrome?
Several of the symptoms of Apert syndrome appear as a result of the malformation of the skull which affects hearing, vision, cognitive ability and respiratory function among others. The following signs are generally observed in individuals born with this disorder:
- Distorted shape of the head and face owing to the abnormal fusion of skull bones. The head has a typically ‘peaked’ appearance, flatter at the back and with a high forehead.
- Sunken and asymmetric face.
- The nose has a bulbous tip.
- The upper jaw is tilted backward with the result that the lower teeth usually project further forward than the upper ones.
- Narrow and arched palate. A cleft palate is present in about 30% of instances.
- A delayed or ectopic eruption of teeth. The teeth are generally positioned close together in the mouth.
- Webbed hands and feet because of the partial or complete fusion of bones in these limbs.
- Thumbs and big toes may be abnormally thick.
- A common nail spanning multiple fingers or toes.
- Bulging, sometimes crossed eyes, set wider apart than usual. The eyelids do not close all the way and tilt downwards. There may be problems with vision.
- Even when born with normal birth weight and length, the individual will eventually display short stature due to the slowing down of growth rate.
- Excessive sweating.
- Brittle nails.
- Excessive wrinkling of forehead skin.
- Severe acne, especially during puberty.
- The fusing of the spinal bones, mainly in the cervical region.
- Impaired intellectual development.
- Noisy breathing and snoring.
- Recurrent ear or sinus infections.
- Hearing impairment.
- Holes in the ventricular walls of the heart.
- The testicles may fail to descend in males and in females, the vagina may be blocked.
What Are The Possible Complications?
Since a number of organs and organ systems can be affected in those born with Apert syndrome, the range of possible complications is accordingly diverse. For instance, the severe buildup of pressure within the skull can eventually result in mental retardation. The eyes, being located in shallow sockets, can develop problems with vision such as blurring. The cleft palate can be responsible for frequent ear infections. Cardiovascular defects such as Fallot’s tetralogy may be present and this is potentially fatal.
What Tests Are Needed To Diagnose Apert Syndrome?
 Apert syndrome can be detected prior to birth with the help of techniques such as fetoscopy or ultrasound. Obtaining fetal DNA samples can help confirm the diagnosis. Although it is possible, such prenatal diagnosis is not common.
Apert syndrome can be detected prior to birth with the help of techniques such as fetoscopy or ultrasound. Obtaining fetal DNA samples can help confirm the diagnosis. Although it is possible, such prenatal diagnosis is not common.
The physical characteristics of this condition are usually clearly apparent at birth and aid in a prompt diagnosis. The abnormal skull development typical of this condition is also observed in other disorders such as Crouzon’s syndrome, Carpenter syndrome, Pfeiffer syndrome, Jackson-Weiss syndrome and Saethre-Chotzen syndrome. However, the fusion of bones in the hands and feet is distinctive of Apert syndrome and can assist in distinguishing it from the other diseases mentioned above.
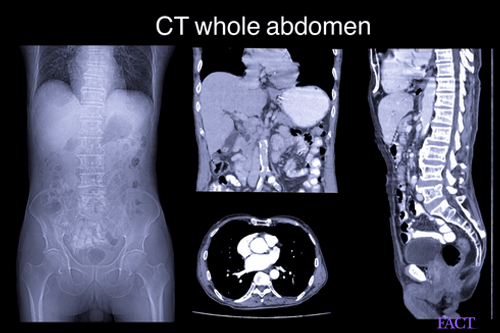
Genetic testing helps confirm the diagnosis. X-rays, computerized tomography (CT) and magnetic resonance imaging (MRI) can help further understand skeletal abnormalities in the skull, vertebrae, hands, and feet as well as congenital heart defects. Psychometric and hearing assessment must also be conducted.
Treatment and Prevention
Treatment – Apert syndrome has no known cure. Surgery to correct the abnormal connection between bones is the main treatment for Apert syndrome.
- Craniosynostosis release (release of skull bone fusion). Here surgery is done to separate the abnormally fused skull bones and partially rearrange some of them. This is done when the child is between 6-8 months.
- Midface advancement – As the child with Apert syndrome grows, the facial bones again become misaligned. Surgery is done by cutting the bones in the cheeks and jaw to bring forward into a more normal position. This surgery is done when the child is between 4-6 years-old.
- Hypertelorism correction (correction of wide-set eyes). This surgery is done to remove a wedge of bone in the skull between the eyes. The surgeon brings the eye sockets closer and may adjust the jaw.
Prevention – There is no current known prevention of Apert syndrome.
- Individuals who have Apert syndrome may meet with genetic counselors to learn about the risk of having children with the disorder.
- If one parent has Apert syndrome, there is a 50% chance of passing it on to their child.
- Prenatal screening and diagnosis may be done to determine if a mother’s fetus has the Apert syndrome.
- https://rarediseases.org/rare-diseases/apert-syndrome/
- https://ghr.nlm.nih.gov/condition/apert-syndrome
- https://rarediseases.info.nih.gov/diseases/5833/apert-syndrome
- http://www.seattlechildrens.org/medical-conditions/chromosomal-genetic-conditions/apert-syndrome/
- http://www.wch.sa.gov.au/services/az/divisions/psurg/craniofacial/documents/apert_syndrome.pdf
Dos and Don'ts
Dos
- Make sure that your child is breathing well as an immediate priority.
- Ensure that your child has proper dental care and bite alignment.
- Provide your child with proper medication and treatment from doctors specializing in Apert syndrome.
Don'ts
- Neglect your child’s mental health as it this disease can make children feel segregated from others.
- Neglect any authentic and necessary surgery.
- Ignore the possibilities of related disorders.
Help Others Be Fit
Related Conditions



Trending Topics








































































































































About
Related Conditions


Subscribe to free FactDr newsletters.
REVAMP YOUR
LIFE
HEALTH
WELLNESS

If you're enjoying our website, we promise you'll absolutely love our new posts. Be the first one to get a copy!
Get factually correct, actionable tips delivered straight to your inbox once a week.
We hate spam too. We will never share your email address with anyone. If you change your mind later, you can unsubscribe with just one click
By clicking Subscribe, I agree to the FactDr Terms & Conditions & Privacy Policy and understand that I may opt out of FactDr subscriptions at any time.
×
How can we improve it?
×
Happy to know you loved our article!
Did it give you information that you used / can use in your life?

