
Thalassemia
Last Updated December 20th, 2021
Thalassemia – Quick Facts
- Annually there are over 332,000 children born worldwide with hemoglobin disorders. About 56,000 of them suffer from some major thalassemia. This includes 30,000 who would need regular blood transfusions and 5500 who would die perinatally of thalassemia major.
- Over 9 million carriers become pregnant annually. The risk that their partner is also a carrier ranges from 0.1-40% (14% average globally). In principle, all couples need to be informed and given the offer of partner testing.
- The name thalassemia was coined from two separate Greek words “thalassa” meaning sea and “emia” meaning blood. Thalassemia means “sea in the blood.” It was coined by George Whipple and William Bradford at the University of Rochester in upstate New York.
- It generally affects people from the Mediterranean and also parts of Southeast Asia, the Middle East, and Africa.
What is Thalassemia?
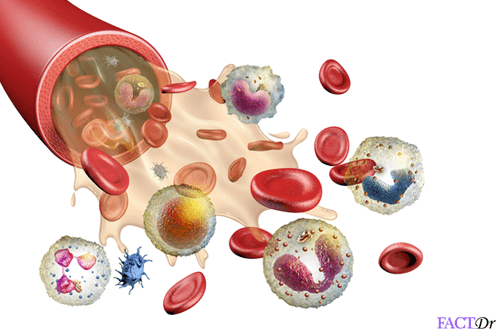
Thalassemia is a blood disorder in which the body makes an abnormal form or does not make enough quantity of the protein hemoglobin. Since hemoglobin is the protein in the red blood cells that carries oxygen, thalassemia can lead to a shortage of oxygen in the blood (anemia). This happens through the destruction of red blood cells in the blood.
Thalassemia is an inherited disorder, which means that if you must get it, at least one of your parents must be a carrier. It is caused by mutation or deletion of key fragments in the genes.
There are different types of thalassemia. The treatment methods and the severity of the disorder will depend on the kind of thalassemia you have. They could be alpha or beta thalassemia and further classified as minor and major depending on the severity.
What is Mediterranean Anemia?
 The gene for beta thalassemia is not evenly distributed amongst the people of the world. It is particularly common in Italy and Greece (even Turkey), all of which are countries around the Mediterranean. This is why it is also sometimes referred to as Mediterranean anemia.
The gene for beta thalassemia is not evenly distributed amongst the people of the world. It is particularly common in Italy and Greece (even Turkey), all of which are countries around the Mediterranean. This is why it is also sometimes referred to as Mediterranean anemia.
General Symptoms of Thalassemia
The signs and symptoms you experience depend on the type and severity of your condition. Some babies show signs and symptoms of thalassemia at birth, while others may develop them during the first two years of life. Some people who have only one affected hemoglobin gene don’t experience any thalassemia symptoms. Some general symptoms of thalassemia are:
- Fatigue
- Pale or yellowish skin
- Dark urine
- Weakness
- Abdominal swelling
- Rapid heartbeat rate
- Cramps
- Cold hands and feet
- Shortness of breath
- Slow growth
- Facial bone deformities
Causes of Thalassemia
Thalassemia is an abnormality or defect in one of the genes that are responsible for the production of hemoglobin. Thalassemia is inherited by the child from its parents.
If only one of your parents has thalassemia, then you might inherit a minor form of the disorder called thalassemia minor, which does not have any symptoms. It is harmless and does not require treatment.
If both of your parents are carriers of thalassemia, then you are at a greater risk of developing a serious form of thalassemia. This condition requires treatment.
Types of Thalassemia
 The normal adult hemoglobin consists of two parts: heme and globin. The globin part is comprised of four chains: two identical alpha globin chains and two identical beta globin chains.
The normal adult hemoglobin consists of two parts: heme and globin. The globin part is comprised of four chains: two identical alpha globin chains and two identical beta globin chains.
Beta Thalassemia
In beta thalassemia, the beta globin chains are affected and body has a problem producing beta globin. It could be classified in to two types depending on the extent of the disorder.
Minor
In thalassemia minor, the children would have one copy of the beta thalassemia gene and a normal beta chain gene. Such children will have only a slight lowering of hemoglobin in their blood. It very closely resembles a mild anemia (iron deficiency). Their red blood cells are smaller. However, their blood iron levels are still normal unless they are iron deficient because of other reasons. This means that no treatment is necessary for this mild type of thalassemia. Iron supplementation is neither helpful nor advised by doctors.
Major
A child born with thalassemia major has two genes with beta thalassemia and no normal beta-chain gene. The clinical picture associated with beta thalassemia major was first described in 1925 by Thomas Cooley. Hence, it is also called Cooley’s anemia.
At birth, the baby with thalassemia major will seem very normal. This is because at birth a baby will have what is called fetal hemoglobin (HbF). It consists of two alpha chains and two gamma chains. There are no beta chains. Therefore, beta thalassemia major does not affect the baby then.
Within the first few months after birth, the baby shows signs of anemia. These include reduced feeding, problems with growth, fatigue, and bouts of fever with diarrhea and other intestinal problems.
Beta thalassemia major can lead to death resulting from heart complications as early as 30.
Alpha Thalassemia
Alpha thalassemia occurs when the body cannot make alpha globin. It happens when one or more of the four genes that control the making of the alpha globins is (are) defective or affected.
This type of thalassemia is of two serious types: hemoglobin H disorder and hydrops fetalis.
Hemoglobin H disorder results when a person is missing three alpha globin genes or is experiencing a change in them. This can lead to bone issues. This can lead to overgrowth of bones in the cheeks, jaws and forehead. Additionally, it can cause:
- Jaundice
- Malnourishment
- Extreme enlargement of the spleen
Hydrops fetalis develops when all four alpha globin genes are altered or missing. It is a rare and severe kind of alpha thalassemia. It occurs before birth. Most babies affected by this condition die shortly after birth or are stillborn.
The milder forms of alpha thalassemia are caused by:
- 1 mutated gene. You will have no symptoms of the disorder. However, you are a carrier of the disorder and can pass it on to your children. Therefore, you are considered a silent alpha thalassemia carrier.
- 2 mutated genes.Symptoms are usually mild. Here, you are considered to have an alpha-thalassemia trait.
Risk Factors for Thalassemia
Family history of thalassemia
Thalassemia is passed from parents to children through mutated hemoglobin genes. If you have a family history of thalassemia, you may have an increased risk of the condition.
Certain ancestry
Thalassemia occurs most often in African-Americans and in people of Mediterranean and Southeast Asian ancestry.
Complications of Thalassemia
Possible complications of thalassemia are:
- Iron overload.There is a build-up of iron in the bodies of thalassemia patients because of the disorder itself and because of blood transfusions. This iron overload can cause damage to the heart, liver, and the endocrine system.
- People with thalassemia are at an increased risk of acquiring infection. This is especially true in people who have had their spleen removed.
In cases of severe thalassemia, the following complications can occur:
 Bone deformities. Thalassemia causes your bone marrow to expand. This leads to an abnormal bone structure, especially in the face and head. This expansion of the bone marrow makes the bones thin, brittle, and susceptible to breakage.
Bone deformities. Thalassemia causes your bone marrow to expand. This leads to an abnormal bone structure, especially in the face and head. This expansion of the bone marrow makes the bones thin, brittle, and susceptible to breakage.- Enlarged spleen (splenomegaly).The spleen helps your body fight infection. It also filters out unwanted blood cells. Since thalassemia destroys a large number of red blood cells, your spleen is enlarged and needs to work harder than normal. This can not only make anemia worse but also reduce the life of transfused red blood cells.
- Heart problems.Congestive heart failure and abnormal heart beat rhythms can result from thalassemia.
- Slowed growth rates.It can slow down the growth of a child and delay puberty.
Thalassemia and Pregnancy
Thalassemia affects reproductive function in pregnant women. It can lead to infertility. You must plan ahead of time to ensure the health of your baby. If you wish to conceive, your iron levels will be monitored. The doctor will also monitor you closely if you have any pre-existing organ damage. Nowadays, there are even assistive reproductive techniques that help you conceive a normal baby even if you have thalassemia. This involves genetically modifying the embryo.
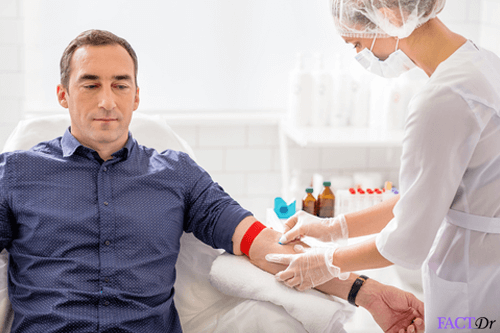
Diagnosis of Thalassemia
Most children with moderate to severe thalassemia show signs and symptoms within their first two years of life. If your doctor suspects your child has thalassemia, he or she may confirm a diagnosis using blood tests. Usually, beta-thalassemia screening is performed in both prenatal and antenatal cases, if there is risk of this genetic disorder being inherited by the baby.
If your child has thalassemia, blood tests may reveal:
- A lower level of red blood cells
- Red blood cells that are varied in size and shape
- Red blood cells that are smaller in size than expected
- Pale red blood cells
- Red blood cells with uneven hemoglobin distribution, which lends a bull’s-eye appearance to the cells when viewed under the microscope
Blood tests may also be used to:
- Evaluate his or her hemoglobin
- Measure the amount of iron in your child’s blood
- Perform DNA analysis to diagnose thalassemia or if the person is merely a carrier of mutated hemoglobin genes.
Prenatal testing
Testing for thalassemia can be done before a baby is conceived. Tests used to diagnose thalassemia in fetuses include:
- Chorionic villus sampling.It is done around the 11th week of pregnancy and it involves removing a tiny piece of the placenta for further testing.
- This test is usually done around the 16th week of pregnancy. It involves taking a sample of the fluid that surrounds the fetus.
Treatment for Thalassemia
Treatments for moderate to severe thalassemia may include:
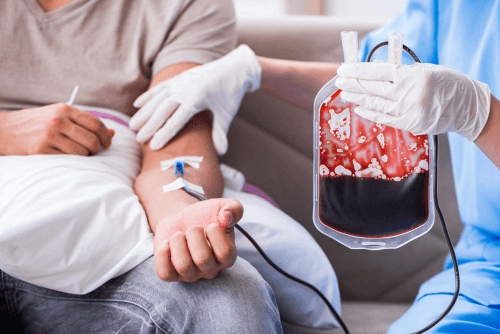
- Frequent blood transfusions.More severe forms of thalassemia require frequent blood transfusions. Over time, these transfusions can increase the iron content in your blood. This will adversely affect the functioning of your heart, liver and endocrine system. To combat the iron overload the doctor might prescribe some medicines to lower your iron levels.
- Stem cell transplant.Also called a bone marrow transplant, a stem cell transplant may be the solution for severe cases of thalassemia. This will ensure that lifelong transfusions and treatments for iron overload in the system are not required. During this procedure, you will receive the infusion of stem cells from a donor, mostly a sibling.
Home remedies for managing thalassemia
In addition to sticking to your treatment plan for thalassemia, the following tips will help manage the disorder.
- Avoid excess iron.Do not take supplements that contain iron unless your doctor recommends them.
- Eat a healthy diet. Eat a balanced diet rich in vitamin D and calcium to help rebuild your bones. You might have to take folic acid supplements as well to increase the number of red blood cells. Keep your doctor informed of your diet. Also, consult with him/her if there are any other supplements that you must take.
- Avoid infections. Protect yourself from infection by frequently washing your hands with soap before and after food. You will also need annual flu shots and vaccines for meningitis, hepatitis B to prevent infection. In case you develop a fever or are exhibiting the signs and symptoms of an infection, contact your doctor immediately for treatment.
Final Words
Thalassemia is a disorder of the blood hemoglobin (that leads to anemia in some cases) and is inherited from parents who also carry it. Thalassemia minor is mildly symptomatic and does not generally require treatment. Thalassemia major, on the other hand, is severe and requires continuous treatment. Thalassemia can be quite fatiguing, but you don’t have to be alone in the fight.
Several thalassemia support groups are out there and joining them can help you with strategies for managing the disorder. The life expectancy of a person with thalassemia depends on the kind of thalassemia, the severity of symptoms, and the treatment he/she receives.
https://www.healthline.com/health/thalassemia#outlook
http://sickle.bwh.harvard.edu/hbsynthesis.html
https://www.cdc.gov/ncbddd/thalassemia/facts.html
https://www.medicinenet.com/beta_thalassemia/article.htm
https://ghr.nlm.nih.gov/condition/beta-thalassemia
https://www.who.int/bulletin/volumes/86/6/06-036673/en/
https://www.aafp.org/afp/2009/0815/p339.html
https://ada.com/conditions/thalassemia/
https://medlineplus.gov/ency/article/000587.htm
https://www.mayoclinic.org/diseases-conditions/thalassemia/symptoms-causes/syc-20354995
Help Others Be Fit
Related Conditions


Trending Topics































































































































About
Related Conditions


Subscribe to free FactDr newsletters.
REVAMP YOUR
LIFE
HEALTH
WELLNESS

If you're enjoying our website, we promise you'll absolutely love our new posts. Be the first one to get a copy!
Get factually correct, actionable tips delivered straight to your inbox once a week.
We hate spam too. We will never share your email address with anyone. If you change your mind later, you can unsubscribe with just one click
By clicking Subscribe, I agree to the FactDr Terms & Conditions & Privacy Policy and understand that I may opt out of FactDr subscriptions at any time.
×
How can we improve it?
×
Happy to know you loved our article!
Did it give you information that you used / can use in your life?

