
Rett syndrome

Last Updated December 20th, 2021
Overview of Rett syndrome
Neurological disorders are common in different age groups, genders, and demographic groups and may result from abnormalities in the central and peripheral nervous system. A large number of these disorders may result from abnormalities of the central and the peripheral nervous systems. Studies have shown that some of these problems are congenital and may interfere with the communicating abilities of an individual. Rett syndrome is one such disorder which mostly affects females but is also found in males.
According to the current global statistics, 1 in every 10,000 females born is found to have Rett syndrome. The current research works are being directed towards finding the exact cause of Rett syndrome and how to prevent the occurrence of the disease.
What is Rett syndrome?
Rett syndrome, also known as cerebroatrophic hyperammonemia is a rare genetic disorder of the grey matter of the brain. The disease was first described by Andreas Rett in the year 1966. It is usually present at birth and is more common in females, although in a few cases males are also diagnosed with the disease. It is marked by certain clinical characteristics like small hands and feet and a retarded growth of the head. The newborns may exhibit acute microcephaly in some cases.
Some of the aberrant behavioral patterns observed are hand movements like wringing or constantly putting the hands inside the mouth. Up to 80% of these people experience seizures and 50% of the patients are incapable of walking. These children lack verbal communication skills and exhibit certain developmental and gastrointestinal disorders. Rett syndrome has symptoms similar to that of autism, cerebral palsy and Angelman syndrome.
What are the possible causes of Rett syndrome?
Studies have established Rett syndrome as an outcome of mutations in the MECP2 gene located on the X chromosome. In around 10% of the Rett syndrome cases, mutations in the CDKL5 and FOXG1 genes have been found to be responsible for Rett syndrome. The underlying factors of Rett syndrome are discussed below-
- Sporadic mutations: In 95% of the sporadic mutations, the mutated MECP2 is assumed to be derived from a de novo mutation on the male copy of the X chromosome. The exact cause behind the mutation of the sperm is still a matter of ongoing research. Both the parents are genotypically normal and are not carriers of the gene.
- Germline mutations:Here the defective genes are inherited from the mothers who are phenotypically normal, but have a germline mutation in the gene encoding methyl-CpG-binding protein 2 (MECP2). The inheritance, in this case, occurs in the X-linked dominant pattern and is found mainly in the females. The males affected in this case die in the pre-natal stage itself. It is important to mention in this case that an atypical form of Rett syndrome may occur due to a mutation in the gene encoding cyclin-dependent-kinase-like 5 (CDKL5). This type is presented with spasms and early-onset epilepsy and affects nearly 1 in every 12,500 females within the age of 12 years.
- Pontien noradrenergic deficits: A significantly low concentration of norepinephrine is found in the brains of the Rett syndrome patients. A reduction in the tyrosine hydroxylase, which is essentially the rate-limiting enzyme is found to be accountable for this.
- Midbrain dopaminergic disturbances: A major dysregulation of the nigrostriatal pathway in the midbrain is responsible for Rett syndrome in a number of cases studied so far.
What are the main signs and symptoms of Rett syndrome?
 Many of the signs and symptoms of Rett syndrome are similar to that of Autism or Asperger’s. Hence the symptoms are mainly classified as autistic symptoms and non-autistic symptoms. The autistic symptoms of Rett syndrome are discussed below-
Many of the signs and symptoms of Rett syndrome are similar to that of Autism or Asperger’s. Hence the symptoms are mainly classified as autistic symptoms and non-autistic symptoms. The autistic symptoms of Rett syndrome are discussed below-
- Urinary and bowel incontinence
- Screaming and fits
- Constant crying
- Tendency to hyperventilate, hold breath or swallow excess air
- Avoidance of eye-contact
- Absence of reciprocation at social or emotional level
- Tendency to use non-verbal communication during social interactions
- Sensory disorders
- Partial or complete absence of speech (in some cases speech may develop very late)
The non-autistic symptoms of Rett syndrome include the ones that are seen in cerebral palsy or Angelman syndrome. These are discussed below-
- Short stature
- Abnormal body proportions
- Difficulty walking
- Low muscle tone (hypotonia)
- Learning to walk very late
- Abnormalities in gait
- Uncoordinated muscle movements
- Retarded growth of head (microcephaly)
- Gastrointestinal disorders
- Muscular spasticity
- Spasmodic and involuntary movement of the hands and facial muscles (chorea)
- Sustained and repetitive muscle contractions (dystonia)
- Jaw clenching and teeth grinding (bruxism)
What are the different types of Rett syndrome?
In addition to the classical forms of Rett syndrome, several atypical forms of Rett syndrome have been identified through research studies. These are described below-
- Congenital variant: It is also known as Rolando variant and is a severe form of Rett syndrome. The head circumference is abnormal right from the birth. However these patients do not exhibit the characteristic gaze of the Rett syndrome patients.
- Zappella variant:It is relatively milder form of Rett syndrome and is also called the “preserved speech variant”. Here the patients partially acquire the manual skills and language after the age of 5, i.e when the regression phase is completely over. The height, weight and the head circumference of the head are found to be more or less normal.
- Hanefield variant:It is also called the early epilepsy variant, wherein the patients have epileptic fits before five months of age.
Diagnosis and treatment
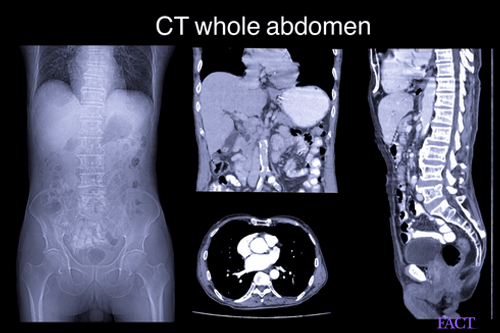
 MRI and CT scans can detect the neurological and structural abnormalities of the brain. Doctors usually recommend the following for easing the complications of Rett syndrome-
MRI and CT scans can detect the neurological and structural abnormalities of the brain. Doctors usually recommend the following for easing the complications of Rett syndrome-
- Selective Serotinin Reuptake Inhibitors
- Anti-psychotics
- Beta-blockers
- Occupational, speech and physical therapy
A large number of the affected population die in the fetal stage itself due to the associated complications of Rett syndrome. The children who survive should be kept under intensive care and treatment in order to reduce the risks of co-morbidities in future.
- http://www.nichd.nih.gov/health/topics/rett
- https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Rett-Syndrome-Fact-Sheet
- https://www.ninds.nih.gov/Disorders/All-Disorders/Rett-Syndrome-Information-Page
- https://rarediseases.org/rare-diseases/rett-syndrome/
- http://brainfoundation.org.au/disorders/rett-syndrome
Dos and Don'ts
Dos
- Lifelong support system and care are required for patients with Rett syndrome.
- A baby born with Rett syndrome will show normal growth until 6-18 months. It is important to keep a check on the onset of symptoms (mild to severe) so that the condition is detected as early as possible.
- For those patients with Rett syndrome, who develop scoliosis, weight-bearing exercises are good for keeping muscles healthy.
- Refer to a good physiotherapist to help the patient cope up with reduced motor functions.
Don'ts
- Consume an unhealthy diet, lacking in essential nutrients. The diet of the child should help in weight gain since healthy weight levels are closely linked with better social skills and interactions.
- Avoid medication or schooling activities. Anti-seizure medications may be prescribed. Proper therapy can help many patients attend school and improve social skills.
Help Others Be Fit
Related Conditions





Trending Topics








































































































































About
Related Conditions

Subscribe to free FactDr newsletters.
REVAMP YOUR
LIFE
HEALTH
WELLNESS

If you're enjoying our website, we promise you'll absolutely love our new posts. Be the first one to get a copy!
Get factually correct, actionable tips delivered straight to your inbox once a week.
We hate spam too. We will never share your email address with anyone. If you change your mind later, you can unsubscribe with just one click
By clicking Subscribe, I agree to the FactDr Terms & Conditions & Privacy Policy and understand that I may opt out of FactDr subscriptions at any time.
×
How can we improve it?
×
Happy to know you loved our article!
Did it give you information that you used / can use in your life?

