
Sickle cell disease
Last Updated December 20th, 2021
Overview of sickle cell disease
In the recent years, the statistics of Global Genes have shown that there are nearly 7000 rare diseases in the world and an estimated 300 million people live with those diseases. They have also established the fact that 6% of the babies are born with rare genetic diseases and about 30% of them die before the age of five. Sickle cell disease is a serious genetic disorder, causing Anaemia or similar conditions in the patients. It is a hereditary blood disorder and mainly affects the African Americans.
The statistics of the American Society of Haematology show that sickle cell disease affects nearly 8% of the African Americans. Around 5% of the world population are carriers of the gene for sickle cell disease, though they do not have the disease themselves. The current global burden of sickle cell disease and blood disorders of different types is quite high and needs immediate attention.
What is sickle cell disease?
 Sickle cell disease is a group of hereditary blood disorders that arise from mutations in the hemoglobin gene. This hemoglobin is the oxygen carrier present in red blood cells (RBCs). The widely observed form of sickle cell disease is Sickle cell anemia. The Red Blood Cells acquire a peculiar sickle-like shape in this disease. Sickle cell disease may occur as early as 5-6 months of age. The usual complications observed are- anemia, swollen feet and hands, bacterial infections and in the extreme cases, stroke.
Sickle cell disease is a group of hereditary blood disorders that arise from mutations in the hemoglobin gene. This hemoglobin is the oxygen carrier present in red blood cells (RBCs). The widely observed form of sickle cell disease is Sickle cell anemia. The Red Blood Cells acquire a peculiar sickle-like shape in this disease. Sickle cell disease may occur as early as 5-6 months of age. The usual complications observed are- anemia, swollen feet and hands, bacterial infections and in the extreme cases, stroke.
Complications may worsen with age. These people usually do not live more than 40 years. A very interesting feature of this disease is that the patients are protected from malaria, a mosquito-borne disease common in the equatorial countries. The disease is more prevalent among the African Americans. However, a few other ethnic groups such as the inhabitants of the Middle-east, Central part of India and the countries on the borders of the Mediterranean sea like Italy and Greece.
What causes Sickle cell disease?
Sickle cell disease mainly arises when both parents are the carriers of the defective hemoglobin gene. The patient inherits two copies of the mutated hemoglobin gene, one copy from each parent. The proper manifestations of the disease usually occur in the presence of certain triggers like stress, dehydration, temperature and weather changes and high altitude.
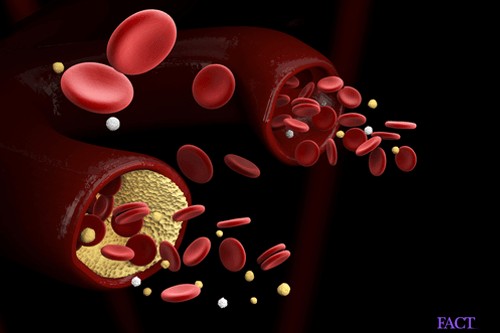
The main complications of the disease are attributable to the progressive loss of elasticity of the Red Blood Cells. Under normal conditions, the healthy Red Blood Cells are very elastic and flexible. This allows the cells to pass through the capillaries of varying width.
In sickle cell disease, there is an acute shortage of oxygen in the body, which is pathologically termed as “low oxygen tension”. This leads to the sickle-like deformations of the Red Blood Cells. This phenomenon occurs repeatedly and this reduces the elasticity of the Red Blood Cells. Even when the normal oxygen conditions are restored, the cell shapes do not go back to normal. Therefore they are unable to deform while passing through the capillaries. This gives rise to ischemia and occlusion of blood vessels.
At a more advanced stage, the red blood cells get destroyed, which is known as hemolysis. This gives rise to the anemic symptoms. The rate of formation of new Red Blood Cells from the bone-marrow becomes much less than the rate of destruction of the existing RBCs. The sickle cells perish much earlier than the normal Red Blood Cells (within 10-20 days). This gives rise to the major complications.
What are the main symptoms of Sickle Cell disease?
 The patients of Sickle Cell Disease mainly exhibit anemic symptoms in the initial stage. With the gradual progress of the disease, the following symptoms are observed-
The patients of Sickle Cell Disease mainly exhibit anemic symptoms in the initial stage. With the gradual progress of the disease, the following symptoms are observed-
- Sickle cell crisis: It begins as anemia. The associated complications (crises) are- aplastic crisis, hemolytic crisis, sequestration crisis, and vaso-occlusive crisis. It usually lasts for 5-7 days.
- Vaso-occlusive crisis: In this condition, the sickle cells obstruct the capillaries and impede the circulation of blood to a particular organ. This results in ischemia, pain, necrosis and eventually organ damage.
- Splenic sequestration crisis: The spleen is affected in many patients owing to its constricted vessels and its function of filtering and discarding the Red Blood Cells.
- Acute chest syndrome: The main symptoms observed are- pulmonary infiltration, chest pain, fever and hypoxemia (oxygen deficiency).
- Aplastic crisis: This condition is triggered by the Parvovirus B19, which attacks the RBC precursors and prevents the formation of Red Blood Cells. In this stage, the patients have baseline anemia, rapid heart rate, pallor, and fatigue.
- Hemolytic crisis: The hemoglobin drop rate, in this case, is very high. It is commonly observed in patients with G6PD deficiency.
What are the main types of sickle cell disease?
The classification of sickle cell disease is done based on the inheritance pattern and the characteristics. These are as follows-
- Sickle cell anemia: Here, the shape of the RBCs is determined by the two defective hemoglobin types that are inherited.
- Sickle Haemoglobin C disease: It is a relatively milder form of sickle cell disease. Here the baby inherits two distinct abnormal hemoglobins- hemoglobin S and C.
- Sickle Beta-Zero disease: The two main diseases under this category are- Sickle Beta-Zero Thalassemia and Sickle Beta-Plus Thalassemia.
- Haemoglobin C disease: It is characterized by a minor abnormality in hemoglobin and is mainly observed in the African American people.
Diagnosis
The following diagnostic tests are mandatory in order to detect the presence of sickle cell disease-
- Complete blood count
- Reticulocyte count
- Sickle solubility test
- Hemoglobin electrophoresis
- Gel electrophoresis
- High-performance liquid chromatography
- Genetic testing (done in the prenatal phase)
Sickle cell disease detected in the gestation period can be cured to a certain extent through the umbilical cord blood transplant. In newborns or in very small children bone marrow transplant is often carried out in order to increase the life expectancy.
Treatment, management, & prevention
Stem cell transplant can potentially cure sickle cell anemia. However, the risks involved are very high which may include death. Thus, it is usually performed in individuals lesser than 16 years of age as above this age group the risks involved increases. Additionally, finding a donor is a tough task.
The management of sickle cell anemia usually focuses on avoiding emergencies, preventing complications and relieving symptoms. As per the guidelines of the Centers for Disease Control and Prevention (CDC), infants and children (up to the age of 2 years) suffering from sickle cell anemia should frequently visit doctors; children more than 2 years of age should visit at least once in a year.
Some of the treatment modalities are:
- Antibiotics: Infants suffering from sickle cell anemia need to be given antibiotics from 2 months of age. The antibiotic course continues up to around 5 years of age. This may keep the infant from life-threatening infections such as pneumonia. In case of adults, if the spleen has been removed or if there is a history of pneumonia, penicillin has to be administered lifelong.
- Pain-relieving medications: These can be given to overcome pain in case of a sickle cell crisis.
- Hydroxyurea: It can reduce the requirement for blood transfusions and hospitalizations and the frequency of painful episodes if taken on regular basis. It stimulates the synthesis of fetal hemoglobin (the kind of hemoglobin present in newborn babies which functions to prevent the formation of sickle cells. It is contraindicated during pregnancy and might increase the risk of infections in the later part of life. It should only be taken as advised by the doctor.
- Blood transfusion- This procedure increases the number of healthy blood cells in the circulation and thereby reduces the symptoms of anemia.
- Bone marrow transplant– This procedure involves the replacement of affected bone marrow by healthy bone marrow.
Other treatment methods include prevention of infections by vaccinations to prevent infections, surgery to rectify vision problems and removal of the spleen in some of the cases. It is an inherited disease caused by gene mutation. Thus prevention of the disease is not possible.
- https://www.cdc.gov/ncbddd/sicklecell/data.html
- https://ghr.nlm.nih.gov/condition/sickle-cell-disease
- https://www.nhlbi.nih.gov/health-topics/sickle-cell-disease
- https://www.mayoclinic.org/diseases-conditions/sickle-cell-anemia/diagnosis-treatment/drc-20355882
- https://www.healthline.com/health/sickle-cell-anemia
Dos and Don'ts
Dos
- Get regular checkups done. An ultrasound is a painless procedure which can assess the risk of getting a stroke for your child.
- Get vaccinated to prevent infections. Clinical manifestations of infections in patients with sickle cell anemia can be severe. Vaccines such as annual flu shot and pneumococcal vaccine should be administered.
- Take folic acid supplements. Folic acid is essential for the formation of new cells. Intake of a balanced diet with whole grains and lots of colorful fruits and vegetables can provide the optimum amount of vitamins and minerals.
Don'ts
- Expose yourself to extreme temperatures. Exposure to very hot or very cold temperatures might increase the possibility of occurrence of sickle cell crisis.
- Resort to self-medication. Over-the-counter (OTC) medications such as naproxen sodium or ibuprofen can be taken for relieving pain only if advised by the doctor. These might cause damage to the kidneys if not taken in the correct dosage.

Help Others Be Fit
Related Conditions



Trending Topics








































































































































About
Related Conditions
Subscribe to free FactDr newsletters.
REVAMP YOUR
LIFE
HEALTH
WELLNESS

If you're enjoying our website, we promise you'll absolutely love our new posts. Be the first one to get a copy!
Get factually correct, actionable tips delivered straight to your inbox once a week.
We hate spam too. We will never share your email address with anyone. If you change your mind later, you can unsubscribe with just one click
By clicking Subscribe, I agree to the FactDr Terms & Conditions & Privacy Policy and understand that I may opt out of FactDr subscriptions at any time.
×
How can we improve it?
×
Happy to know you loved our article!
Did it give you information that you used / can use in your life?

