
Gaucher Disease
Last Updated December 20th, 2021
Overview
Lysosomal storage disorders are characterized by the absence or deficit of enzymes required for the digestion of molecules such as carbohydrates and fats. This leads to a toxic accumulation of the unmetabolized material. Gaucher disease is the most common lysosomal storage disorder. In this case, the body is unable to metabolize a certain lipid or fatty molecule known as glucocerebroside. Gaucher disease is a hereditary condition and is found to occur with greater frequency within certain specific populations. The range, severity and age of onset of symptoms is highly variable from one individual to the other. Some may even be asymptomatic.
Definition
 Gaucher disease is a rare, hereditary metabolic disorder characterized by the deficit of the enzyme glucocerebrosidase. The function of this enzyme is to metabolize a substance known as glucocerebroside. In its absence, abnormally large quantities of glucocerebroside accumulate within the liver, spleen and bone marrow, disrupting the normal functioning of these organs. There are three main types of Gaucher disease, with types 2 and 3 accounting for only about 5% of known cases.
Gaucher disease is a rare, hereditary metabolic disorder characterized by the deficit of the enzyme glucocerebrosidase. The function of this enzyme is to metabolize a substance known as glucocerebroside. In its absence, abnormally large quantities of glucocerebroside accumulate within the liver, spleen and bone marrow, disrupting the normal functioning of these organs. There are three main types of Gaucher disease, with types 2 and 3 accounting for only about 5% of known cases.
The National Gaucher Foundation reports that approximately 1 in every 450 individuals of Ashkenazi Jewish descent is likely to suffer from some form of Gaucher disease. This disease is significantly rarer in the general non-Jewish population where it is believed to affect 1 in every 40,000 individuals. According to the National Organisation for Rare Disorders, a certain variant of type 3 Gaucher disease is more prevalent in the Norrbotten area of Sweden where it is estimated to affect roughly 1 in every 50,000 individuals. In general, this disease affects men and women in equal numbers.
Causes
The direct cause of Gaucher disease is the insufficiency or absence of the enzyme needed in order to break down the fatty molecule known as glucocerebroside. Research has revealed that this occurs because of a recessive mutation in the gene responsible for the synthesis of the necessary enzyme. As many as 150 mutations have been identified that could potentially give rise to this disease.
Gaucher disease is an ‘autosomal recessive disorder’. This means that an individual can only have this disease if he or she inherits the defective gene from both parents. If a person inherits one normal gene and one defective copy, the normal one overrides the defective one. This person will not themselves suffer the effects of the disease, but they are capable of passing the mutation on to their children. Such people are known as carriers. It is estimated that as many as 1 in 10 individuals among those of Ashkenazi Jewish heritage may be carriers of Gaucher disease. When both parents are carriers, the child has a 25% chance of suffering from the disease.
Symptoms
The exact nature of symptoms, their intensity, and age of onset varies depending on the particular type of Gaucher disease. This condition can even be asymptomatic in some individuals. Typical symptoms include:
- Abnormal enlargement of the liver and/or spleen due to the buildup of glucocerebroside.
- Resultant distention of the abdomen.
- Nosebleeds, bleeding from gums and other signs of difficulty in clotting of blood. This results from the malfunctioning of the spleen which causes it to destroy healthy platelets.
- Anemia resulting from the lack of red blood cells.
- Extreme fatigue.
- Enlarged lymph nodes.
- Short stature and growing deficit.
- Severe bone pain, the death of bone tissue because of insufficient blood supply to the bones.
- Osteopenia and osteoporosis.
- Respiratory disorders.
- A variety of neurological symptoms such as seizures and ataxia may be seen in types 2 and 3.
Types of Gaucher disease
There are three recognized types of Gaucher disease.
- Type 1 Gaucher disease: This is the most prevalent, implicated in approximately 95% of known cases. Onset often occurs in childhood. Symptoms can vary widely. However, common indications include abnormal spleen enlargement, deficiency of blood corpuscles and debilitating bone defects. It is known as the non-neuronopathic type because there are generally no neurological symptoms.
- Type 2 Gaucher disease: This type is also known as acute neuronopathic Gaucher disease. It mainly occurs in newborns and young babies, severely affecting the brain. Hence, the risk of early death is very high. Symptoms can even manifest in utero and affected individuals may die prior to birth or soon afterward.
- Type 3 Gaucher disease: This is known as chronic neuronopathic Gaucher disease. Onset usually occurs in childhood or teenage. This type affects the blood, skeletal system and nervous system. Lung disease may also be observed. Life expectancy is fairly low, with patients not known to survive beyond the fourth decade of life.
Some infants suffering from symptoms characteristic of type 2 may manage to survive into early childhood. This is treated as an intermediate type Gaucher disease since it does not fully conform either to type 2 or type 3.
Clinical Complications
 Gaucher disease is associated with an elevated risk of developing malignancies such as liver cancer and various forms of blood cancer.
Gaucher disease is associated with an elevated risk of developing malignancies such as liver cancer and various forms of blood cancer.
The adverse effects of this disease on the skeletal system can produce severe arthritis and even weaken the bones to the extent of developing spontaneous fractures.
Reduced ability of the blood to form clots due to the deficit of platelets increases the chances of internal bleeding which can be fatal. Patients and even carriers of Gaucher disease are more likely to develop Parkinson’s disease later in life.
Generally, early onset of symptoms such as anemia and low platelet count is associated with a poorer outlook. A heightened risk of mortality is particularly pronounced with regard to types 2 and 3.
Diagnosis
Informing one’s physician about a known family history of Gaucher disease can expedite the process of diagnosis. However, in parts of the world where it is rarely encountered, arriving at an accurate diagnosis of Gaucher disease may be challenging. The wide variance in clinical presentation can further complicate the process.
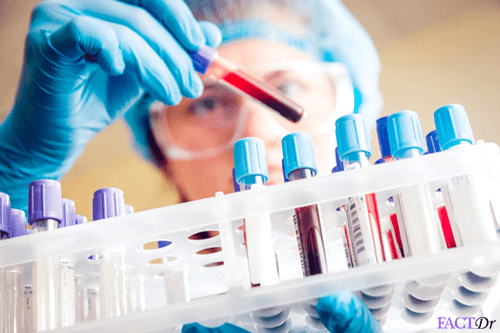
A simple and effective laboratory test is usually sufficient in order to identify Gaucher disease but most physicians and diagnostic laboratories may not be aware of this. This is a blood test known as beta-glucosidase leukocyte or BGL test. Genetic testing confirms the presence of mutations associated with this disease. This is useful in early diagnosis or screening of individuals with a known family history of Gaucher disease. An early diagnosis can be helpful in preventing or containing severe symptoms with potential long-term implications.
Treatment & Prevention
There is no complete cure for Gaucher’s Disease. These various treatment methods help in controlling the various symptoms associated with this disease:
- Medications: Drugs are prescribed that help in enzyme replacement. Medications that hinder fat production and accumulation such as Zavesca are also helpful. Since osteoporosis is a common outcome of Gaucher’s Disease, medicines that help rebuild the bones are also a part of the treatment.
- Surgery: Surgery in case of Gaucher’s Disease is done either for bone marrow transplant or removal of the spleen. A bone marrow transplant can help in reversing the Gaucher’s Disease. Spleen removal is usually the last resort when none of the other methods show an improvement in the condition of the patient.
Since it’s a genetic disorder, there are no specific preventive measures for the same. Genetic testing and counseling can help patients with Gaucher’s Disease take up a call when thinking of starting a family.
Dos and Don'ts
Dos
- Find a physician who is a specialist in Gaucher’s Disease and maintain a detailed list of symptoms you experience.
- Be in touch with your doctor and track the symptoms closely. Adjust the medications only after consulting the doctor.
- Consume a diet rich in Vitamin D and calcium to strengthen your bones and muscles.
Don'ts
- Delay in the treatment of Gaucher’s disease as it can lead to irreversible and serious consequences.
- Let go of your health. Exercise regularly and consume a nutritious and well-balanced diet.
- Ignore genetic counseling before trying to start a family, if you or your partner has a family history of Gaucher’s Disease.
Help Others Be Fit
Related Conditions


Trending Topics









































































































































About
Related Conditions
Subscribe to free FactDr newsletters.
REVAMP YOUR
LIFE
HEALTH
WELLNESS

If you're enjoying our website, we promise you'll absolutely love our new posts. Be the first one to get a copy!
Get factually correct, actionable tips delivered straight to your inbox once a week.
We hate spam too. We will never share your email address with anyone. If you change your mind later, you can unsubscribe with just one click
By clicking Subscribe, I agree to the FactDr Terms & Conditions & Privacy Policy and understand that I may opt out of FactDr subscriptions at any time.
×
How can we improve it?
×
Happy to know you loved our article!
Did it give you information that you used / can use in your life?

